
Overview of uPEPperoni
(The background section of Crowe, Wang and Rothnagel, BMC Genomics 2006, 7:16 offers an excellent lead-in to the following).
uPEPperoni was designed to assist in the location and identification of upstream open reading frames (uORFs) that have the potential to encode bioactive peptides (uPEPs). In order to facilitate quick identification of conserved uORFs, it generates "heatmaps" that allow for visual comparison of pairs of sequences for regions of localised sequence similarity. As a result, uPEPperoni is divided into two subprograms. Conserved uPEP Search allows for the identification of conserved uORFs and uses the full functionality of uPEPperoni, while the Heatmap Generation utility generates heatmaps from any user-entered pair of sequences, and will show regions of localised sequence similarity in an arbitrary pair of nucleotide sequences.
The two subprograms allow for entry into the uPEPperoni pipeline at different stages (Figure 1). The Conserved uPEP Search takes user-entered query sequences and compares them using BLAST to a database of eukaryotic uORFs derived from the RefSeq mRNA release datafiles. It then generates heatmaps based on the alignment of query/BLAST hit (reference) transcript pairs. The Heatmap Generation allows entry at this point, with user entered query/reference sequences. If the locations of the uPEP and coding sequence (CDS) are known for both transcripts, Ka/Ks ratios for both regions are calculated. Both subprograms will accept a user-entered plain sequence of nucleotides ('AATGCGATAGC...', for example), or the RefSeq accession/GI identifier of a query mRNA (eg. 'NM_003463' or 'GI:62865860').
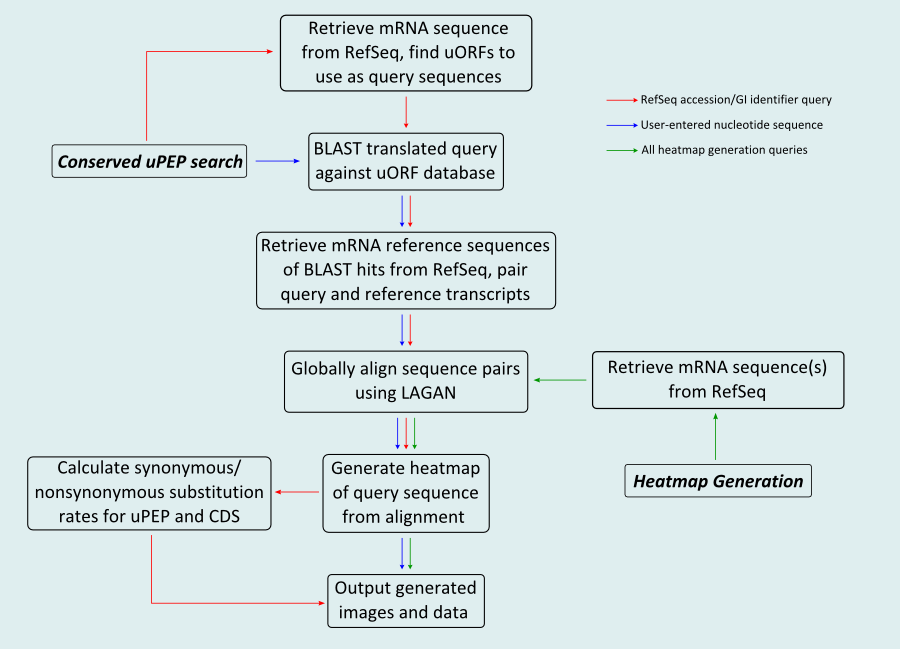
Full descriptions of the settings of each subprogram as well as examples of each are given in the respective subprogram's documentation below.
The Conserved uPEP Search utility (Figure 2) is the default subprogram of uPEPperoni and is displayed when the uPEPperoni main site is first loaded. It can also be accessed by selecting the Conserved uPEP Search tab located on the grey menu bar below the uPEPperoni logo.
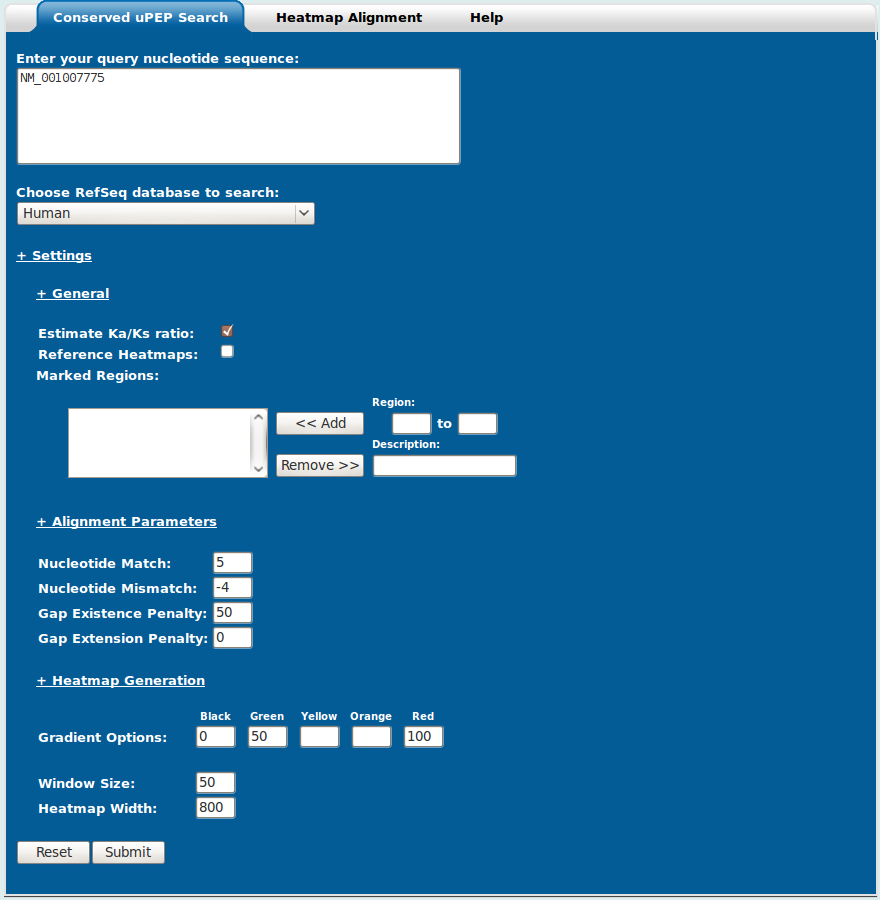
The Conserved uPEP Search form contains a textbox for entering a query sequence (the RefSeq accession 'NM_001007775' has been entered in Figure 2), a combo box for selecting the internal uORF reference databases (the human uORFs database has been selected in Figure 2), and an expandable settings panel. The settings panel can be expanded/collapsed by clicking the "+ Settings" hyperlink and furthermore, each subsection can be expanded/collapsed by clicking on the "+ General", "+ Alignment Parameters", "+ Heatmap Generation" text hyperlinks.
Settings - General:
Calculate Ka/Ks ratio: Checking this option will ask uPEPperoni to estimate synonymous and nonsynonymous substitution rates using the method of Yang and Nielsen (2000), implemented in a library compiled from modified source code of the yn00 program in the PAML package (Yang 2007). The integration of this library into uPEPperoni was done with the permission of the author of PAML, Prof. Ziheng Yang.
Generate Reference Heatmaps: The default behaviour of uPEPperoni after sequence pair alignment is to generate a heatmap representation of the query transcript. However, in certain situations, it is preferable to have a heatmap representation of the BLAST hit or reference sequence (one such example is in the Conserved uPEP Search documentation - Examples section). Selection of this option will generate heatmaps of both sequences involved in an alignment.
Marked Regions: The marked regions section allows you to specify domains and give them descriptions. At the moment, the descriptions aren't added to the final heatmap, so they are only useful to keep track of what has been entered. To specify a region/domain type in the start and end nucleotides in the two boxes below the small "Region:" caption. A description of the region may be entered in the box below the "Description:" caption. Press the "<< Add" button to add the domain. You can remove added domains by selecting them (use Shift or Ctrl to select multiple: Shift for ranges, Ctrl to select multiple individuals), and clicking the "Remove >>" button. The domains will show up as black bars on the heatmap.
Settings - Alignment:
Standard parameters of any sequence alignment. The "Nucleotide Match:" and "Nucleotide Mismatch:" parameters specify the reward and penalty for nucleotide matches/mismatches, while "Gap Existence Penalty:" and "Gap Extension Penalty:" specify the penalties for the opening and extension of gaps in the alignment.
Settings - Heatmap Generation:
Gradient Options: The gradient options allow users to modify the reference colour (heat) gradient from which heatmaps are generated. Placing a value beneath a colour associates that colour with the percentage sequence identity of the value, and colours for inbetween values are linearly interpolated. Threshold values can be created by using values other than 0 and 100 at the extremes.
Window Size: This value allows users to change the size of the window used to to calculate the percentage sequence identity surrounding a nucleotide. Smaller values give greater resolution, while large values allow overall trends to be seen.
Heatmap Width: This option specifies the width (in pixels) of the heatmap uPEPperoni produces.
Conserved uPEP Search - Examples
Examples of usage for both the Conserved uPEP search and Heatmap Generation utilities can be found here.
The Heatmap Generation utility (Figure 3) can be accessed by selecting the Heatmap Generation tab located on the grey menu bar below the uPEPperoni logo.
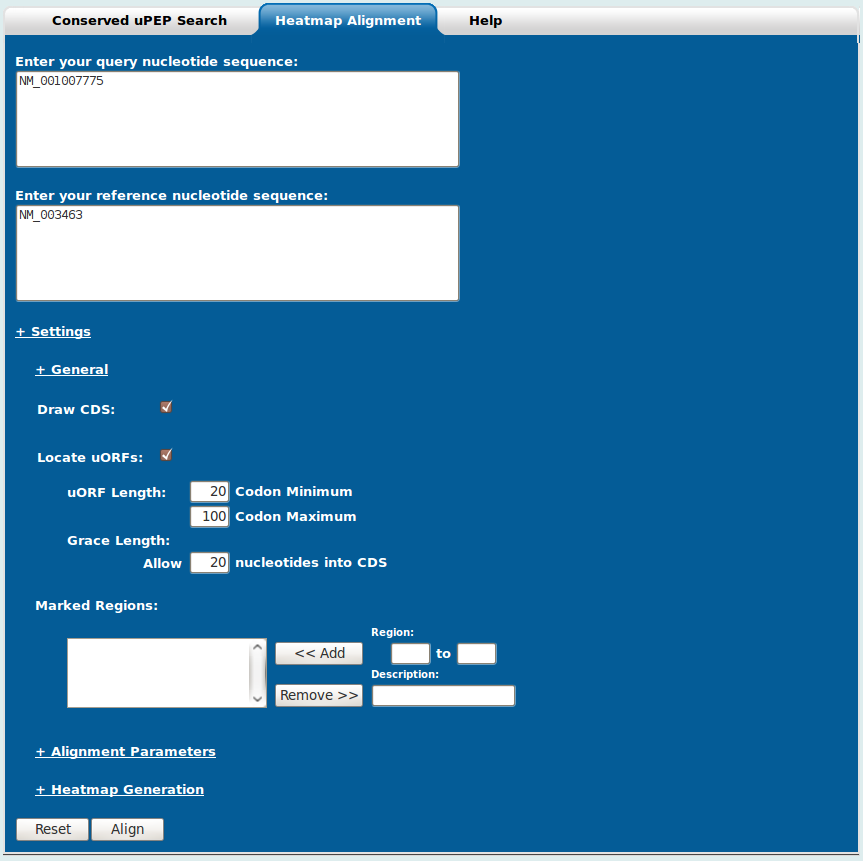
The Heatmap Generation form contains two textboxes, one each for the query and reference sequences and an expandable settings panel. The settings panel can be expanded/collapsed by clicking the "+ Settings" hyperlink and furthermore, each subsection can be expanded/collapsed by clicking on the "+ General", "+ Alignment Parameters", "+ Heatmap Generation" text hyperlinks.
Settings - General:
Draw CDS: If a RefSeq accession query is given, the coding sequence (CDS) of the query sequence will be shown on the final heatmap.
Locate uORFs: If a RefSeq accession query is given, any uORFs matching the entered parameters will be shown on the final heatmap. The parameters allow the user to modify the minimum and maximum size of a uORF as well as how many nucleotides into the CDS a uORF is allowed.
Marked Regions: The marked regions section allows you to specify domains and give them descriptions. At the moment, the descriptions aren't added to the final heatmap, so they are only useful to keep track of what has been entered. To specify a region/domain type in the start and end nucleotides in the two boxes below the small "Region:" caption. A description of the region may be entered in the box below the "Description:" caption. Press the "<< Add" button to add the domain. You can remove added domains by selecting them (use Shift or Ctrl to select multiple: Shift for ranges, Ctrl to select multiple individuals), and clicking the "Remove >>" button. The domains will show up as black bars on the heatmap.
Settings - Alignment:
Standard parameters of any sequence alignment. The "Nucleotide Match:" and "Nucleotide Mismatch:" parameters specify the reward and penalty for nucleotide matches/mismatches, while "Gap Existence Penalty:" and "Gap Extension Penalty:" specify the penalties for the opening and extension of gaps in the alignment.
Settings - Heatmap Generation:
Gradient Options: The gradient options allow users to modify the reference colour (heat) gradient from which heatmaps are generated. Placing a value beneath a colour associates that colour with the (sequence) percentage identity of the value, and colours for inbetween values are linearly interpolated. Threshold values can be created by using values other than 0 and 100 at the extremes.
Window Size: This value allows users to change the size of the window used to to calculate the percentage sequence identity surrounding a nucleotide. Smaller values give greater resolution, while large values allow overall trends to be seen.
Heatmap Width: This option specifies the width (in pixels) of the heatmap uPEPperoni produces.
Acknowledgements
The authors of uPEPperoni would like to thank Prof. Ziheng Yang for his permission to include libraries generated from his PAML source code.
References
Yang, Z. 2007. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24: 1586-1591.
Yang, Z., and R. Nielsen. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Molecular Biology and Evolution 17: 32-43.